New Site Launched: Looking for oligos? Visit our new oligo and Stellaris dedicated platform at
oligos.biosearchtech.com
The Flexibility of PNA Synthesis
Introduction
The use of PNA oligos is well known in therapeutic1 and diagnostic applications.2 This arises from the non-natural neutral backbone resulting in its ability to stabilise duplexes and triplexes, thereby enabling the use of shorter probes with higher specificity. In addition, the fact that these oligos are nuclease resistant makes them ideal candidates for therapeutic applications.
There are two types of PNA synthesis:
- Conventional; using established peptide synthesis chemistry
- Panagene’s method using self-activating monomers
Conventional methods have been used since the early 1990s and remain the most widely used method of PNA synthesis.
There are three main protection chemistries used. The most common being Fmoc/Bhoc (LK5001 - LK5004)3, the second (original) being Boc/Z (1 - 4)3 and the third MMT/Base labile protection (5 - 8)4. The structures of these monomers are shown in Figure 1.
Boc/Z and Fmoc/Bhoc chemistries are very much based on peptide synthesis and are both compatible with the synthesis of PNA-peptides. MMT/base labile monomers were specifically designed to be used in the solid phase synthesis of PNA-DNA chimera and are fully compatible with oligo synthesis using phosphoramidite chemistry.
Regardless of the chemistry utilised, there are some design rules that need to be considered. PNA oligos have poor solubility which is exaggerated where the sequence is purine rich, in particular G-rich since this leads to aggregation of the oligo. As with DNA sequences, poly G sequences are known to have strong G:G interactions (cf quadruplex formation). Also, the neutral backbone of PNA results in much stronger duplex stabilisation than the corresponding DNA:DNA duplex therefore it is important to avoid self-complementarity within the sequence. The most efficient synthesis is observed where the following rules are adhered to.
- Short oligos; < 15mers
- Purine content of < 50%
- Avoid self-complementarity
- Avoid polyG sequences
In reality however there these cannot always be applied and there are ways to improve the synthesis and downstream handling of more complex PNA oligos.
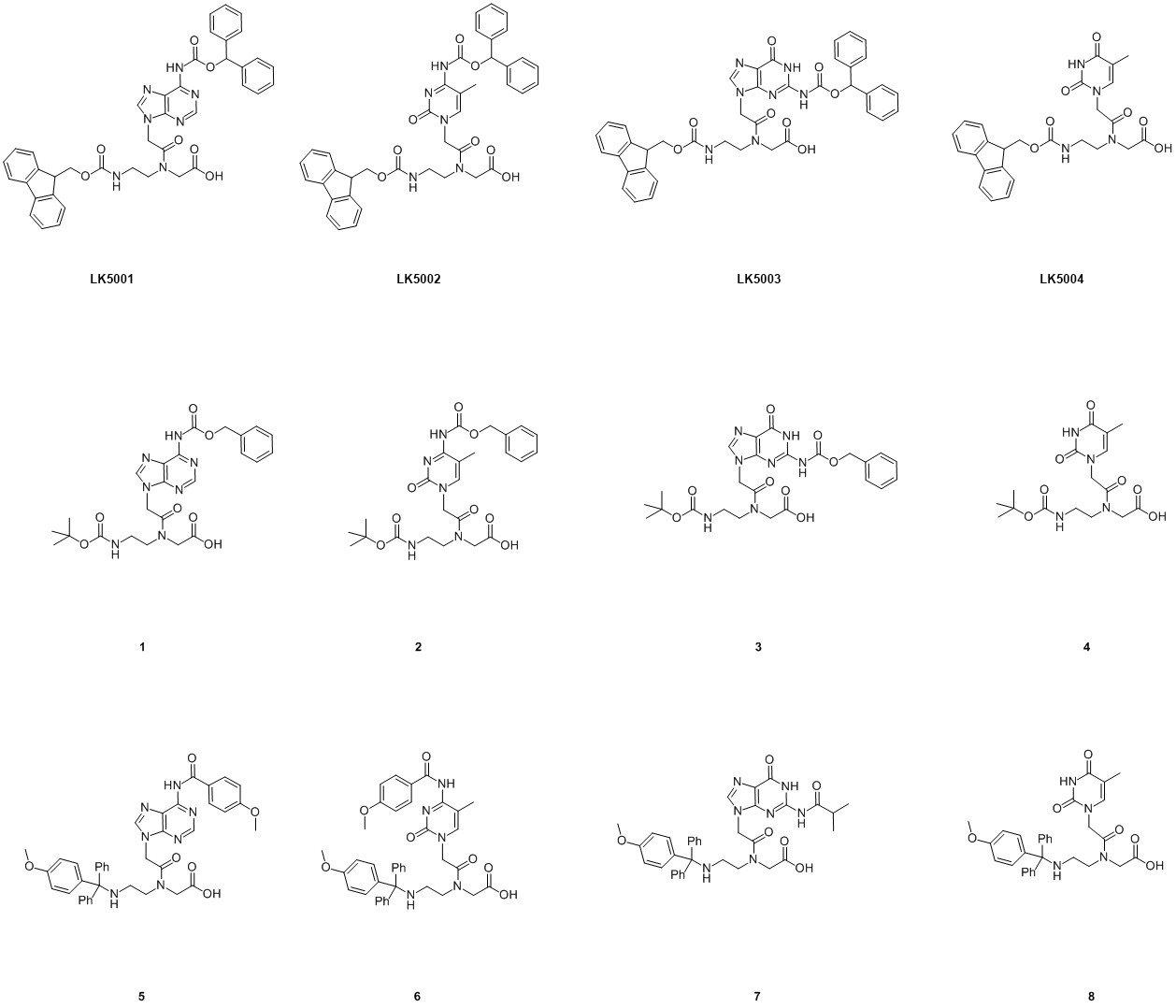
Figure 1. Structures of the three main monomer types used in PNA synthesis. (Click to view full-size image)
PNA Synthesis using Fmoc/Bhoc Chemistry
PNA synthesis using Boc/Z monomers is considered to give oligos with higher purity than using Fmoc/Bhoc monomers since fewer side reactions are observed. However, the use of TFA to remove the Boc group in each synthesis cycle limits this chemistry in terms of its compatibility with other chemistries and, unlike Fmoc/Bhoc chemistry, is not suitable for use on a DNA synthesiser. As a result the most commonly used PNA synthesis chemistry is Fmoc/Bhoc. The general PNA synthesis cycle is shown in Figure 2.
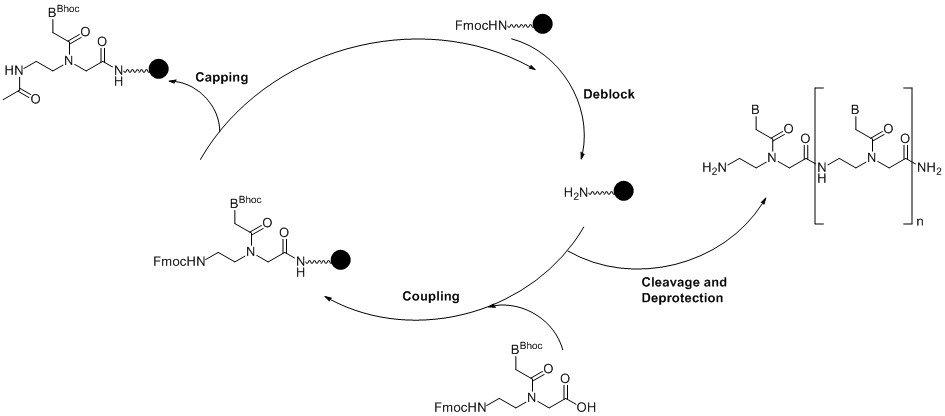
Figure 2. Solid phase PNA synthesis cycle. (Click to view full-size image)
PNA synthesis on DNA synthesisers such as Expedites is well known, but scale is limited, as are the number of ports for modified PNA monomers or amino acids (for the synthesis of PNA-peptides). As a result many users have transferred this chemistry to peptide synthesisers and, where the instrument allows for microwave assisted synthesis, the cycle time is rapidly decreased. In either case, a universal solid support is used; typically XAL-PEG-PS or PAL-PEG-PS which are commonly used in peptide synthesis.
The oligo is synthesised from the C-terminus (pseudo 3’) just as DNA is synthesised from the 3’-end of the oligo. The first step in the cycle removes the Fmoc protection from the support using 20% piperidine in DMF or NMP (N-methyl pyrrolidinone) followed by coupling of the first monomer.
In general the monomer is dissolved in NMP. DMF is also feasible but often contains trace amount of amines therefore will reduce the lifetime of the monomer solution. The coupling reaction is activated with a peptide coupling reagent such as HATU in the presence of a base, typically 2,6-lutidine.
As with DNA synthesis, although the coupling efficiency is high there is a need to cap any unreacted sites to prevent deletions in the sequence. The synthesis returns to the beginning of the cycle and the Fmoc group is removed from the N-terminus.
As mentioned previously, Fmoc/Bhoc monomers can lead to some minor side reactions. These are base-catalysed rearrangements that occur during Fmoc deprotection. The mechanisms are shown in Figure 3. However, these are very minor products and have been measured at 0.3-0.4% contamination using a 2 minute deprotection time. This is 4 times the deprotection time used in the synthesis cycle on an Expedite and >4 times where a microwave assisted peptide synthesiser is used.
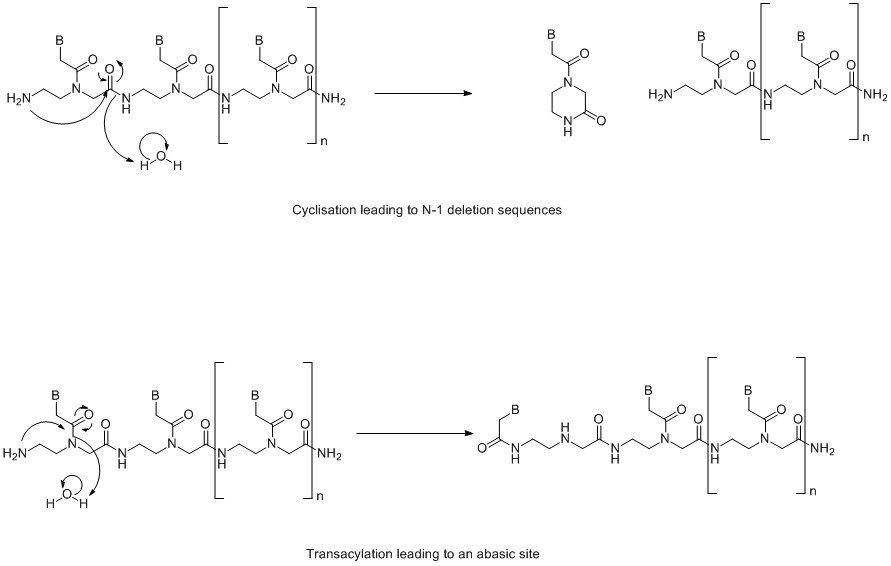
Figure 3. Base catalysed rearrangement reactions. (Click to view full-size image)
As with the 5’-DMT group in DNA synthesis, the Fmoc group can be removed prior to cleavage and deprotection or retained to assist with HPLC purification. Cleavage and deprotection is carried out with 20% m-cresol in TFA, although, depending on the amino acids used in PNA-peptide synthesis, other scavengers may be required. The oligo is then isolated by precipitation with diethyl ether, ready for purification. This is typically carried out by RP-HPLC on C18 column using 0.1%TFA in water / 0.1%TFA in MeCN as the mobile phase.
PNA-peptide synthesis follows the same synthesis cycle and, provided there are sufficient ports on the synthesiser, it is possible to synthesise the entire sequence in one run. The amino acids clearly have to be compatible with Fmoc/Bhoc PNA synthesis therefore Fmoc(Bhoc) amino acids are generally the first choice of reagent. Other amino acid chemistries also include Fmoc(Boc), Fmoc(pfb), Fmoc(tOBu), Fmoc(Trt), Fmoc(tBu) and Fmoc(Mtt). It is often advantageous to include at least one spacer but more typically three, between the PNA and peptide sequences. This most commonly used spacer is the AEEA spacer (LK5005) or linker (also known as eg1 linker) as shown in Figure 4.
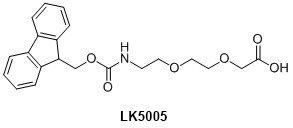
Figure 4. AEEA linker structure.
It is well known that PNA oligos tend to have low aqueous solubility and that the introduction of 2-3 AEEA linkers (or in combination with Lys residues) has been shown to improve this.
Labelling PNA and PNA-peptides
There are many reasons to label PNA or PNA-peptides.5 Such uses include a fluorescent marker on a PNA probe for use in FISH, or a delivery reagent such as cholesterol to aid cellular uptake, although for the most part cell penetrating peptides are employed for the latter.6
The nature of the label will dictate whether the conjugation is carried out on-column or post synthetically. Provided the label is stable to the cleavage and deprotection conditions required, on-column labelling is generally more efficient.
On-Column Labelling
The most obvious method is to modify the N-terminus where the Fmoc is removed during oligo synthesis and add the label using either the NHS ester of the label or the free acid and a peptide coupling reagent such as HATU. Typically, at least two AEEA linkers (5005) are added between the N-terminal base or amino acid to improve the efficiency of the conjugation. The main advantage of using on-column labelling is where amino acids with secondary amino side chains (such as Lys) are used, since these are protected during the on-column conjugation. In fact by using orthogonal protection in these sites it is possible to add multiple labels or even to incorporate a branching point to allow further PNA or peptide synthesis. See Figure 5.
In this example, the N-terminus is labelled first, since removal of the Fmoc group is incorporated in the synthesis cycle; the label is added via NHS ester chemistry. Dde protection is then removed from one Lys residue allowing labelling, or in this case branching, at this site. After cleavage and deprotection there is still the opportunity to add an additional label on the remaining Lys residue post-synthetically.
Although the example shows the Lys residues at the N-terminus, these can equally be incorporated at the C-terminus or indeed within the sequence.
Clearly the design rules for PNA have to be considered in synthesising such modified constructs. It is perfectly possible that there may have to be adjustments to the coupling step e.g. double or triple coupling and/or an increased concentration of monomers.
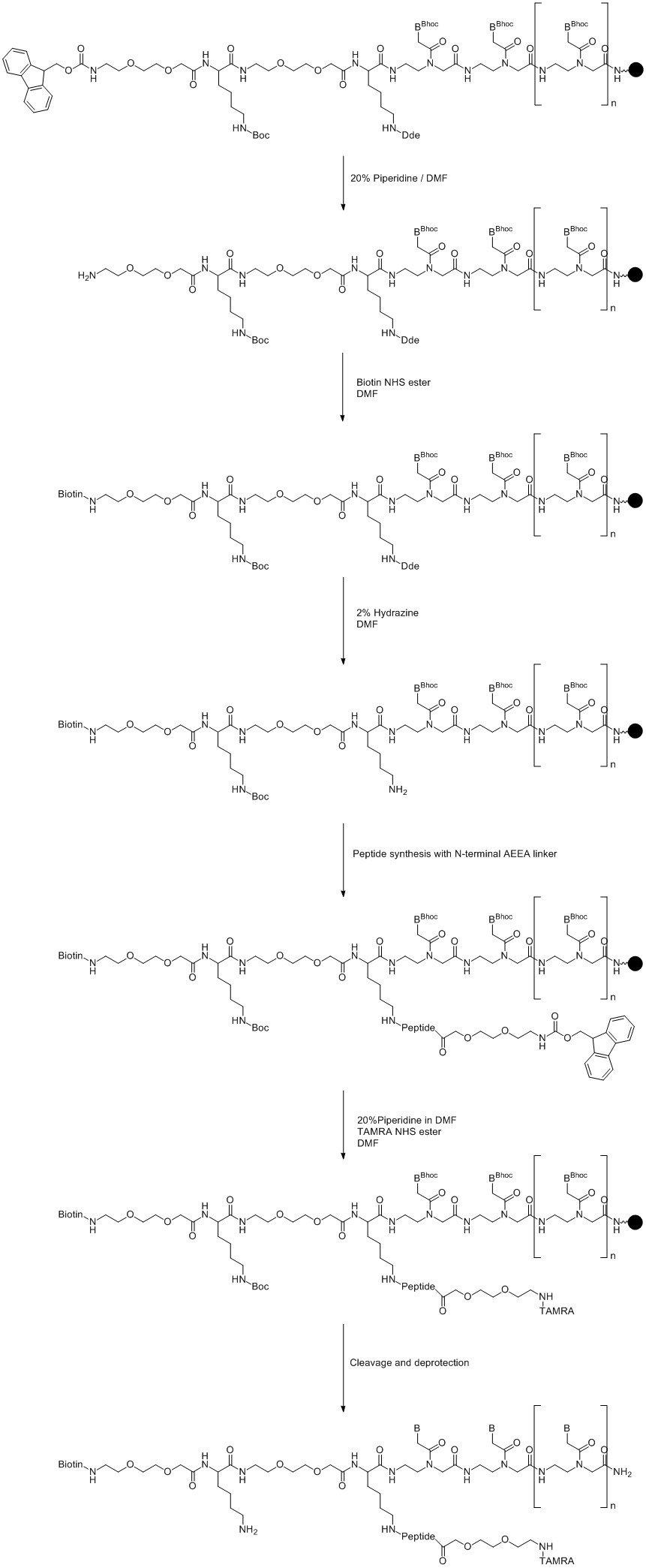
Figure 5. Multiple on-column labelling of Lys modified PNA. (Click to view full-size image)
Also using this approach in conjunction with Cys residues allows further modification where the label is added via a maleimide or haloacetamide.
Post-synthetic labelling
This is generally only carried out where the conjugation label is acid-sensitive or where an amino acid residue does not lend itself to on-column labelling. Unlike DNA, this type of conjugation can be carried out in organic solvents such as DMF, NMP or DMSO; since the backbone is neutral, there is no need for an aqueous buffer and as a result it tends to be more efficient. However, as with DNA, this is dependent on the properties of the oligo itself, e.g. secondary structure, solubility.
As with on-column conjugations, the coupling reaction is more efficient where AEEA linkers (LK5005) are incorporated between the PNA oligo and the label. In general coupling is carried out on the N-terminus with NHS ester forms of the label, but equally Cys residues can be used to couple using maleimide or haloacetamides.
Alternative linkers for PNA conjugation
While the AEEA linker (LK5005) is the most commonly used linker in PNA synthesis, it is possible to use other linkers. At LINK we will soon introduce new thiol linkers for use in PNA synthesis; see Figure 6. These are particularly useful where a label is only reactive with a thiol and the use of Cys residues is not optimal, e.g. where this would interfere with the functionality of a PNA-peptide. These can be incorporated either at the N-terminus or on a branching point incorporated as described above. The thiol linkers are coupled in the same way as LK5005.
Both linkers are suitable for on-column conjugation where LK5010 is deprotected using hydroxylamine or LK5011 using TCEP.
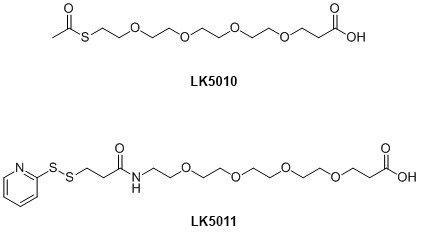
Figure 6. Alternative linkers for PNA conjugation.
Please note that the dPEG® products 5010 and 5011 have been discontinued.
Modified PNA Monomers
The hybridisation properties of PNA are favourable in terms of stabilising a duplex; there are far fewer modified PNA monomers available than for oligonucleotide synthesis, but some do exist. Examples are shown in Figure 7.
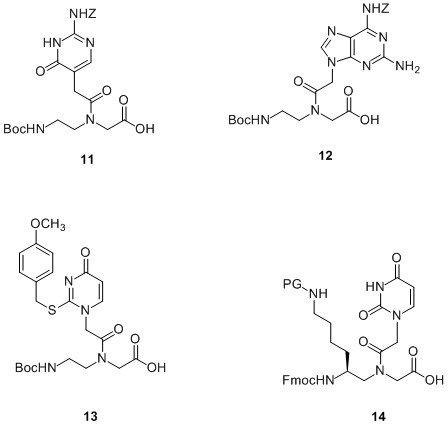
Figure 7. Examples of modified PNA monomers.
The first base modification reported was pseudoisocytosine7 or J-PNA (11) for incorporation into the Hoogsteen strand in the preparation of pH independent bis-PNA for use in triplex formation. This was originally prepared for Boc/Z chemistry but has since been prepared with Fmoc/Bhoc protection.8
2,6-Diaminopurine (DAP) is well known to stabilise duplexes and is often used in DNA and RNA duplexes to achieve this. Incorporation of the DAP-PNA monomer (12) into a PNA sequence9 has been shown to increase the Tm of a duplex by 2.5-6.5oC per incorporation. The specificity when DAP is base pairing against a mismatch is higher than when the mismatch is paired against an A residue. It has also been shown that when it is paired against 2-thioU (13) or 2-thioT, the duplex is destabilised. In fact a decamer of PNA where all the A bases have been replaced with DAP and all T with thioU will hybridise to its complementary DNA sequence better than the unmodified PNA sequence but will not hybridise to the complementary unmodified PNA sequence.10 This is thought to be useful where mixed purine-pyrimidine PNA is required for strand invasion of a DNA double helix utilising only the Watson-Crick base pairing. In this case duplex-forming PNA oligos are required therefore the use of an unmodified sequence used in conjunction with the complementary DAP/thioU modified sequence will allow strand invasion without the complication of PNA:PNA hybridisation.
As mentioned previously, there is often a need to conjugate labels to PNA. However, depending on the position of the conjugation site and the nature of the label, there is the possibility that this can lead to destabilisation of the duplex in comparison with the analogous unlabelled oligo.11 It has been shown that incorporation of a side chain in the gamma position of PNA monomers, such as 14 in Figure 7, resolves this issue.12 Since it is possible to incorporate one gamma-substituted PNA monomer every third residue with no adverse effect on the resulting duplex, the use of orthogonal protecting groups on a side chain would allow for the incorporation of multiple labels within the sequence.
Conclusion
Since the invention of PNA in 1991 by Peter Nielsen and Michael Egholm, many advances have been made to this chemistry in terms of oligo synthesis and development of modifiers to allow the properties of a PNA sequence to be fine-tuned to best fit its intended use. Many commercial applications are now using PNA technology as the basis of their detection system e.g. DestiNA Genomics or DiaCarta’s QClamp, and research using PNA oligos as therapeutic agents is still very active. It is clear that PNA chemistry remains an area of interest within the therapeutic and diagnostic markets.
References
- (a) Peptide nucleic acids as therapeutic agents, P.E. Nielsen, Curr. Opin. Struct. Biol., 9, 353-7, 1999, Abstract;
(b) Recognition of chromosomal DNA by PNAs, K. Kaihatsu, B.A. Janowski and D.R. Corey, Chem. Biol., 11, 749–758, 2004, Full Text HTML;
(c) Antisense properties of peptide nucleic acid, H.J. Larsen, T. Bentin and P.E. Nielsen, Biochim. Biophys. Acta, 1489, 159–166, 1999, Abstract. - (a) Use of DNA and peptide nucleic acid molecular beacons for detection and quantification of rRNA in solution and in whole cells, C. Xi, M. Balberg, S.A. Boppart and L. Raskin, Appl. Environ. Microbiol., 69, 5673–5678, 2003, Full Text HTML;
(b) Cyclopentane-modified PNA improves the sensitivity of nanoparticle-based scanometric DNA detection, J. K. Pokorski, J.-M. Nam, R.A. Vega, C.A. Mirkin and D.H. Appella, Chem. Commun., 2101 – 2103, 2005, Full Text PDF;
(c) PNA molecular beacons for rapid detection of PCR amplicons, E. Ortiz, G. Estrada and P.M. Lizardi, Mol. Cell. Probes, 12, 219–226, 1998, Abstract. - Peptide Nucleic Acids: Protocols & Applications, 2004, Horizon Biosciences, Ed Peter E. Nielsen.
- Synthesis and properties of PNA/DNA chimeras, E. Uhlman, D.W. Will, G. Breipohl, D. Langner and A. Ryte, Agnew. Chem. Int. Ed. Engl., 35, 2632-2635, 1996, Abstract.
- Peptide nucleic acid conjugates: Synthesis, properties and applications, Z.V. Zhilina, A.J. Ziemba and S.W. Ebbinghaus, Curr. Top. Med. Chem., 5, 1119-1131, 2005, Abstract.
- Peptide nucleic acid (PNA) cell penetrating peptide (CPP) conjugates as carriers for cellular delivery of antisense oligomers, T. Shiraishi and P.E. Nielsen, Artificial DNA PNA XNA, 2, 90-99, 2011, Full Text HTML.
- Efficient pH-independent sequence-specific DNA binding by pseudoisocytosine-containing bis-PNA, M. Egholm, L. Christensen, K.L. Dueholm, O. Buchardtt, J. Coull and P.E. Nielsen, Nucleic Acids Research, 23, 217-222, 1995, Full Text PDF.
- New Fmoc pseudoisocytosine monomer for the synthesis of a bis-PNA molecule by automated solid-phase Fmoc chemistry, P. Neuner and P. Monaci, Bioconjugate Chem., 13, 676−678, 2002, Abstract.
- Increased DNA binding and sequence discrimination of PNA oligomers containing 2,6-diaminopurine, G. Haaima, H.F. Hansen, L. Christensen, O. Dahl and P.E. Nielsen, Nucleic Acids Research, 25, 4639–4643, 1997, Full Text PDF.
- Double duplex invasion by peptide nucleic acid: A general principle for sequence-specific targeting of double-stranded DNA, J. Lohse, O. Dahl and P.E. Nielsen, PNAS, 96, 11804-11808, 1999, Full Text HTML.
- Modulation of the pharmacokinetic properties of PNA: Preparation of galactosyl, mannosyl, fucosyl, N-acetylgalactosaminyl, and N-acetylglucosaminyl derivatives of aminoethylglycine peptide nucleic acid monomers and their incorporation into PNA oligomers, R. Hamazavi, F. Dolle, B. Tavitian, O. Dahl and P.E. Nielsen, Bioconjugate Chem., 14, 941–954, 2003, Abstract.
- (a) Gamma-substituted peptide nucleic acids constructed from L-Lysine are a versatile scaffold for multifunctional display, E.A. Englund and D.H. Appella, Angew. Chem. Int. Ed. Engl., 46, 1414-1418, 2006, Abstract;
(b) Chiral peptide nucleic acids with a substituent in the N-(2-Aminoethyl)glycine backbone, T. Sugiyama and A. Kittaka, Molecules, 18, 287-310, 2013, Full Text PDF.
Applicable products
Added to basket
PNA monomers are manufactured and sold pursuant to licence under one or more of US Patents Nos. 5,773,571, 6,133,444, 6,172,226, 6,395,474, 6,414,112, 6,613,873, 6,710,163 and 6,713,602, or corresponding patent claims outside the US. PNA Monomers are sold to be used for internal research use only, and are not to be resold unless by separate licence.
dPEG® products are sold under licence from Quanta Biodesign, Ltd, (Plain City, OH), are sold for laboratory use only and are not intended to be used for any other purposes, including but not limited to, in vitro diagnostic purposes, in foods, drugs, medical devices, cosmetics or commercial use. A separate licensing agreement/supply agreement with Quanta BioDesign, Ltd. is required to use the products in applications beyond laboratory use. Customers are responsible to informing Quanta BioDesign, Ltd when products are being used beyond lab use. dPEG® technology is protected by US Patents #7,888,536 and 8,637,711 and US Patent Pending #2013/0052130. dPEG® is a registered trademark of Quanta BioDesign, Ltd.